โรคเซลล์ประสาทสั่งการเสื่อม
(Motor neuron diseases, MNDs)
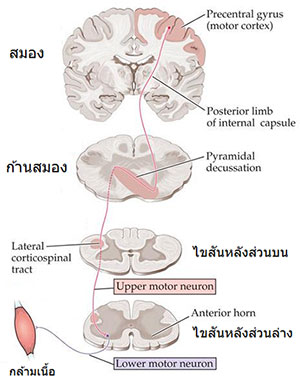
เซลล์ประสาทสั่งการ (Motor neuron) ของคนเรามี 2 ระดับสำคัญคือ
ระดับบน เริ่มจากผิวสมอง ส่งใยประสาทลงมาก้านสมองและไขสันหลัง เรียกว่า Lateral corticospinal tract ระดับล่าง อยู่ที่ Anterior horn ของไขสันหลัง รับสัญญาณจากใยประสาทระดับบนแล้วส่งต่อไปยังกล้ามเนื้อ
หากเซลล์ประสาทสั่งการระดับบนถูกทำลาย (Upper motor neuron lesion, UMNL) จะทำให้กล้ามเนื้ออ่อนแรงแบบแข็งเกร็ง รีเฟล็กซ์ไว เช่นในโรคหลอดเลือดสมองหรือเนื้องอกสมอง ในขณะที่ความผิดปกติของระดับล่าง (Lower motor neuron lesion, LMNL) จะทำให้กล้ามเนื้ออ่อนแรงแบบปวกเปียก อาจมีการสั่นพริ้ว และต่อมากล้ามเนื้อจะลีบฝ่อ เช่นในโรคโปลิโอหรือการบาดเจ็บไขสันหลังรุนแรง
โรคเซลล์ประสาทสั่งการเสื่อมเป็นกลุ่มโรคที่ทำให้เซลล์ประสาทสั่งการที่ควบคุมการเคลื่อนไหวโดยสมัครใจค่อย ๆ ฝ่อและตาย ระบบประสาทอัตโนมัติและการรับความรู้สึกยังปกติ จึงไม่มีผลต่อการมองเห็น การได้ยิน การรับรส กลิ่น หรือความเจ็บปวด และไม่กระทบต่อสติปัญญา ความจำ หรือการทำงานของอวัยวะภายใน สาเหตุของโรคนี้ยังไม่ทราบแน่ชัด จึงยังไม่มีวิธีรักษาให้หายขาด อัตราการดำเนินโรคต่างกันในแต่ละคน บางรายอาจอยู่ได้เพียง 6 เดือน แต่บางรายอยู่ได้นานหลายสิบปี
อาการของโรค
ตำแหน่งเซลล์ประสาทสั่งการสำคัญมี 3 บริเวณ คือ ก้านสมอง (ควบคุมการเคี้ยว กลืน และกล้ามเนื้อใบหน้า), ไขสันหลังส่วนคอ (ควบคุมคอ แขน และกระบังลม ซึ่งเป็นกล้ามเนื้อหลักของการหายใจ), และไขสันหลังส่วนล่าง (ควบคุมลำตัว ขา หูรูดต่าง ๆ)
ดังนั้นอาการจะแตกต่างกันตามตำแหน่งที่เสื่อมและชนิดของเซลล์ประสาทที่ได้รับผล
ตัวอย่างโรคสำคัญในกลุ่ม MND ได้แก่
1. โรคเอแอลเอส (Amyotrophic lateral sclerosis, ALS)
พบมากที่สุดในกลุ่ม MND ประมาณ 1 ใน 20,000 ราย มักเกิดในชาวตะวันตก ร้อยละ 10 มีสาเหตุจากพันธุกรรม (เช่นยีน C9ORF72, SOD1) รอยโรคเกิดทั้งใน Lateral corticospinal tract และ Anterior horn อาจเป็นข้างเดียวหรือทั้งสองข้าง ทำให้กล้ามเนื้ออ่อนแรงทั้งแข็งเกร็งและปวกเปียก มีตะคริวและกล้ามเนื้อเต้น หากโรคลุกลามถึงก้านสมองหรือไขสันหลังส่วนคอ จะเกิดอาการกลืนลำบาก เคี้ยวยาก พูดไม่ชัด และหายใจลำบากจนต้องพึ่งเครื่องช่วยหายใจ
อาการเริ่มหลังอายุ 45 ปีเป็นส่วนใหญ่ แต่ผู้ที่เริ่มตั้งแต่อายุน้อย เช่นกรณีศาสตราจารย์สตีเฟน ฮอว์กิง จะดำเนินโรคช้ากว่าและมีอายุยืนยาวกว่า อาการแรกมักเริ่มจากมืออ่อนแรง หยิบจับของยาก เดินสะดุด เป็นตะคริวบ่อย แล้วค่อย ๆ ลามจนช่วยเหลือตัวเองไม่ได้ ในผู้สูงอายุ บางรายอาจมีอาการคล้ายสมองเสื่อมตามมา แต่ความแตกต่างคืออาการกล้ามเนื้ออ่อนแรงเกิดก่อน
2. โรคพีแอลเอส (Primary lateral sclerosis, PLS)
เกิดจากการเสื่อมของเซลล์ประสาทระดับบนเพียงอย่างเดียว กล้ามเนื้อจึงอ่อนแรงแบบแข็งเกร็ง เริ่มจากขาแข็ง เดินลำบาก แล้วลามไปแขน ลำตัว ใบหน้า และลิ้น ต่างจาก ALS ตรงที่ไม่มีอาการอ่อนปวกเปียกหรือกล้ามเนื้อลีบ และดำเนินโรคช้ากว่า ผู้ป่วย PLS อาจอยู่ได้ถึง 20 ปี และเป็นโรค MND ที่พบน้อยและมีอายุขัยยืนที่สุด
3. โรคเอสเอ็มเอ (Spinal muscular atrophy, SMA)
เกิดจากการเสื่อมของเซลล์ประสาทระดับล่างที่ Anterior horn ของไขสันหลัง สาเหตุจากยีน SMN1 ผิดปกติ ทำให้สร้างโปรตีน SMN ไม่พอ จึงทำให้กล้ามเนื้ออ่อนแรงปวกเปียก รีเฟล็กซ์หาย และกล้ามเนื้อลีบ แบ่งเป็น 5 ชนิดตามอายุที่เริ่มอาการ ตั้งแต่ทารกแรกเกิด (SMA type 0) ที่รุนแรงและเสียชีวิตใน 2 ปีแรก ไปจนถึง SMA type 4 ที่เกิดในผู้ใหญ่และมีอายุขัยใกล้ปกติ
โรคเอสเอ็มเอในเด็ก (4 กลุ่มแรก) มีการถ่ายทอดทางพันธุกรรมแบบ Autosomal recessive พบประมาณ 1:10,000 ของเด็กแรกเกิด ส่วนกลุ่มสุดท้ายเชื่อว่าเกิดจากการกลายพันธุ์ของยีนในภายหลัง
4. โรคบัลบาร์พัลซี (Progressive bulbar palsy, PBP)
เกิดจากการเสื่อมของเซลล์ประสาทระดับล่างที่ก้านสมอง บริเวณเส้นประสาทสมอง IX, X, XII ทำให้กล้ามเนื้อปาก ลิ้น และคอหอยอ่อนแรง เคี้ยวและกลืนลำบาก น้ำลายไหล พูดเสียงขึ้นจมูก ลิ้นสั่นและฝ่อ โรคดำเนินเร็วและมักเสียชีวิตภายใน 3 ปีจากการหายใจล้มเหลว
โรคบัลบาร์พัลซีพบได้ทุกช่วงอายุ ถ้าเกิดในวัยทารกจะเรียกว่า Infantile progressive bulbar palsy ร้อยละ 5-10 มีประวัติคนในครอบครัวเป็นแบบเดียวกัน ในกลุ่มนี้มียีน SOD1 ผิดปกติ
5. โรคซูโดบัลบาร์พัลซี (Pseudobulbar palsy)
เกิดจากรอยโรคที่เซลล์ประสาทระดับบนบริเวณก้านสมอง ทำให้กล้ามเนื้อปาก ลิ้น และเพดานปากแข็งเกร็ง กลืนลำบาก และพูดลำบาก ต่างจากบัลบาร์พัลซีตรงที่ลิ้นไม่สั่นหรือฝ่อ และรีเฟล็กซ์ต่าง ๆ ไวผิดปกติ อีกทั้งมักมีอารมณ์แปรปรวนร่วมด้วย สาเหตุอาจเกิดจาก MND โดยตรง หรือจากโรคหลอดเลือดสมอง เนื้องอก และการบาดเจ็บ
การวินิจฉัย
อาศัยการซักประวัติ ตรวจร่างกาย และคัดออกโรคอื่นที่คล้ายกัน
ลักษณะสำคัญที่บ่งชี้ MND ได้แก่
- กล้ามเนื้ออ่อนแรง 2 ข้าง แต่มักไม่เท่ากันทั้ง 4 แขนขา
- กล้ามเนื้อลีบถ้าเป็นมานานเกิน 6 เดือน
- ไม่มีอาการปวดหรือชา
- ความคิด ความจำปกติ (ยกเว้นระยะท้ายของโรค)
- โรคมีแต่เป็นมากขึ้นเมื่อเวลาผ่านไป (ไม่มีดีขึ้นหรือหายไปบางช่วงเวลา)
การคัดออกโรคอื่นเช่น ตรวจเลือดวัดระดับเอนไซม์กล้ามเนื้อเพื่อแยกโรคของกล้ามเนื้อออกไป ตรวจ MRI สมองและไขสันหลังเพื่อแยกโรคเนื้องอก การติดเชื้อ การบาดเจ็บ การกดทับ และการขาดเลือดของสมองและไขสันหลังออกไป การตรวจน้ำไขสันหลังเพื่อแยกโรคติดเชื้อและมะเร็งกระจายที่เยื่อหุ้มสมองออกไป
การตรวจวินิจฉัยที่สำคัญของโรคเซลล์ประสาทสั่งการเสื่อมคือ
- Nerve conduction velocity (NCV) เป็นการวัดความเร็วในการนำไฟฟ้าของเส้นประสาทที่มาเลี้ยงกล้ามเนื้อที่อ่อนแรง โรคเซลล์ประสาทสั่งการเสื่อม motor nerve ควรจะนำไฟฟ้าได้ช้าลง และมีลักษณะ prolonged distal latencies ซึ่งแสดงว่ามี demyelination ขณะที่ sensory nerve ปกติ
- Electromyography (EMG) เป็นการตรวจคลื่นไฟฟ้าของกล้ามเนื้อที่อ่อนแรง ซึ่งโรคเซลล์ประสาทสั่งการเสื่อมจะให้ลักษณะ spontaneous depolarization at rest (คือกล้ามเนื้อเต้น/สั่นเองขณะพัก) และเมื่อกระตุ้นด้วยไฟฟ้าจะให้ลักษณะ Increased duration, Increased polyphasicity, Increased amplitude, Decreased recruitment, และ Motor unit potential instability
- การตัดกล้ามเนื้อไปตรวจทางพยาธิวิทยา เพื่อแยกโรคของกล้ามเนื้อและรอยต่อของใยประสาทกับกล้ามเนื้อออกไป
ในสถาบันใหญ่อาจตรวจ Motor unit number estimation (MUNE), Transcranial magnetic stimulation (TMS), และ Threshold tracking techniques เพื่อช่วยวินิจฉัยโรคในระยะแรก
การรักษา
ยังไม่มียาที่รักษาให้หายขาด การรักษามุ่งเน้นการประคับประคอง เช่น กายภาพบำบัด ฝึกกลืน ใช้เครื่องช่วยหายใจหรือสายให้อาหาร ยาที่ช่วยยืดอายุได้ เช่น Riluzole (Rilutek®) และ Edaravone (Radicava®) ที่ลดการทำลายเซลล์ประสาทจากกลูตาเมต ในเด็ก SMA อาจใช้ Nusinersen (Spinraza®) เพื่อกระตุ้นการสร้างโปรตีน SMN ยาประคับประคองอื่น เช่น Baclofen, Tizanidine, Botulinum toxin หรือยาต้านซึมเศร้า ก็ใช้เพื่อควบคุมอาการ
พยากรณ์โรค
โรคในกลุ่มนี้มีความรุนแรงและไม่สามารถรักษาให้หายขาด
ALS และ PBP มักมีอายุขัยสั้นที่สุด (หลายเดือนถึงไม่เกิน 5 ปี), PLS มีอายุขัยยาวนานกว่า (ถึง 20 ปี), SMA มีความรุนแรงต่างกันตั้งแต่เสียชีวิตในวัยทารกจนถึงอายุขัยใกล้เคียงปกติในผู้ใหญ่ คุณภาพชีวิตผู้ป่วยขึ้นอยู่กับการได้รับการดูแลและการใช้เทคโนโลยีช่วยชีวิต
การป้องกัน
เนื่องจากยังไม่ทราบสาเหตุที่แท้จริง จึงยังไม่มีวิธีป้องกันโดยตรง
อย่างไรก็ตาม การตรวจพันธุกรรมสำหรับครอบครัวที่มีประวัติ MND โดยเฉพาะ SMA และ ALS แบบพันธุกรรม อาจช่วยลดความเสี่ยงในรุ่นต่อไป การดูแลสุขภาพทั่วไป เช่น การออกกำลังกายพอเหมาะ การหลีกเลี่ยงสารพิษและการติดเชื้อที่กระทบระบบประสาท ก็อาจช่วยลดปัจจัยเสี่ยงบางประการ
สรุป
โรคเซลล์ประสาทสั่งการเสื่อม (MNDs) เป็นกลุ่มโรคที่เกิดจากการเสื่อมของเซลล์ประสาทสั่งการ ส่งผลให้กล้ามเนื้ออ่อนแรง ลีบ และไม่สามารถควบคุมการเคลื่อนไหวได้
แม้จะไม่กระทบต่อการรับรู้และประสาทสัมผัส แต่การดำเนินโรคจะเลวร้ายลงเรื่อย ๆ และยังไม่มียาที่รักษาให้หายขาด ปัจจุบันการรักษามุ่งเน้นการประคับประคองและยืดคุณภาพชีวิตของผู้ป่วยให้ดีที่สุด การทำความเข้าใจโรคนี้ช่วยให้ผู้ป่วยและครอบครัวเตรียมการดูแลและปรับตัวได้เหมาะสม
บรรณานุกรม
- "Motor neuron disease." [ระบบออนไลน์]. แหล่งที่มา Wikipedia. (24 กันยายน 2568).
- "Amyotrophic Lateral Sclerosis (ALS) Fact Sheet." [ระบบออนไลน์]. แหล่งที่มา NIH. (24 กันยายน 2568).
- "Amyotrophic lateral sclerosis." [ระบบออนไลน์]. แหล่งที่มา Wikipedia. (24 กันยายน 2568).
- "Spinal muscular atrophy." [ระบบออนไลน์]. แหล่งที่มา NIH. (24 กันยายน 2568).
-
Anuradha Duleep, Jeremy Shefner. 2013. "Electrodiagnosis of Motor Neuron
Disease." [ระบบออนไลน์]. แหล่งที่มา Phys Med Rehabil Clin N Am 24 (2013) 139–151. (24 กันยายน 2568).